Kidney cancer
Epidemiology: Kidney cancer (renal tumor) accounts for roughly 3 percent of all cancers. the incidence has been rising steadily in Europe and the United States for the past 3 decades, with a particular rise in the proportion of small, asymptomatic tumors detected incidentally via abdominal imaging. Worldwide, approximately 150,000 people are diagnosed with renal cell carcinoma, resulting in 78,000 deaths annually. In the United States, it is estimated that 58000 people are found having kidney cancer and 13000 patients die from this disease each year.
Evidence of geographic variation exists, with the highest incidences occurring in northern Europe and North America and the lowest occurring in Asia and Africa.
Renal cell carcinoma occurs nearly twice as often in men as in women. Higher rates are reported for black versus white Americans, and for urban compared with rural populations.
The average age at diagnosis of RCC is 60-64 years. However, 7% of sporadic RCC is diagnosed in patients youger than 40 years, and rare cases have been reported in patients aged 14-18 years without evidence of familial disorders.
Cigarette smoking doubles the likelihood of renal cell carcinoma and contributes to as many as one-third of cases. Obesity is also a risk factor, particularly in women. Other risk factors include hypertension, unopposed estrogen therapy, and occupational exposure to petroleum products, heavy metals, or asbestos (Motzer et al., 1996). For prevention and treatment of kidney cancer, click here.
Kidney cancer types: There are four types of kidney tumors: renal cell tumor or renal cell carcinoma (RCC), Wilms' tumor, transitional cell tumor (TCT) or reanl transitional cell carcinoma (TCC) or renal urothelial carcinoma (UC), and renal sarcoma. Of them, Renal cell tumor or RCC is the most common type of kidney cancer (>80%).The Heidelberg histologic classification of renal cell tumors subdivides renal cell tumors into benign and malignant parenchymal neoplasms and, where possible, limits each subcategory to the most common documented genetic abnormalities (Kovacs et al., 1997). Malignant tumors are subclassified into common or conventional renal cell carcinoma (clear cell, CCRCC); papillary renal cell carcinoma (PRCC); chromophobe renal cell carcinoma (CRCC); collecting duct carcinoma (CDC), with medullary carcinoma of the kidney; and unclassified renal cell carcinoma. The common or conventional type accounts for about 75% of renal cell neoplasms and is characterized genetically by a highly specific deletion of chromosome 3p. Papillary renal cell carcinoma accounts for about 10% of renal cell tumors. Chromophobe renal cell carcinoma accounts for approximately 5% of renal cell neoplasms. The unclassified renal cell carcinoma represents 5%. Genetically, chromophobe RCC is characterized by a combination of loss of heterozygosity of chromosomes 1, 2, 6, 10, 13, 17, and 21 and hypodiploid DNA content. Collecting duct carcinoma accounts for about 1% of renal cell carcinoma. In children, the Wilms' tumor is the most common type. Benigh tumors includes oncocytoma, renal papillary adenoma, renal fibroma or hamartoma, and angiomyolipoma.
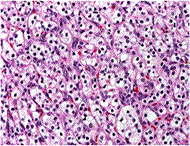
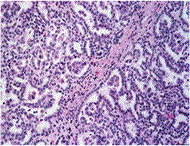
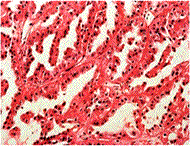
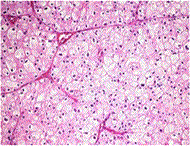
Symptoms: Kidney cancer is most often characterized by blood in the urine, severe and unrelenting pain on one side of the back or abdomen, lumps in the abdomen, and lower back pain that isn't caused by injury. Other symptoms include rapid and unexplained weight loss, recurring fever, edema and loss of appetite.
Etiology (Risk factors): Smoking, obesity, and hypertension are the 3 most well-established risk factors associated with development of sporadic RCC. Smoking is calculated to play a role in the development of RCC in 10-20% of cases among women and 20-30% of cases among men. Obesity is calculated to contribute to the development of 30% of cases in Europe and up to 40% in the United States and Canada. Relative risks for RCC reported for hypertension range from 1.3-2.0. By contrast, only 1-5% of RCC in contemporary surgical series are associated with recognized hereditary genetic disorders.
Genetics:
(1) Gene alterations/modifications which mainly associated with UMPP and HIF pathways or chromotin remodeling. Theses genes include VHL, PBRM1, TP53, KDM5C/JARID1C, BAP1, SETD2, TSC1, NF2, BHD, FH, c-MET, c-MYC, KDM6A/UTX, AHNAK, SHANK1, FAM111B, ASB15, CUL7, SYNE2, CSMD3, ZNF804A, LRP1B, AKAP13, SPTBN4, RYR1, NAV3, CARD11, LRRK2, FMN2, BTRC, APC, PTEN, ARID1A, NORE1, LSAMP, FITH, and so on. The most mutated genes are VHL and PBRM1 and both of them are associated with clear cell RCC.
(2) Cytogenetic abnormalities (e.g. numeric and structural changes such as gain of chromosome 7, 17, 8, 12, 16, 20, X; loss of Y chromosome, chromosome 3p loss; chromosome 3-related translocations; gain of chromosome 5q) are involved in kidney cancer. Chromosome 3-related translocations usually involve CCRCC. Chromosome 3-related translocations include t(1;3)(q32.1,q13.3), t(1;3)(q41;q27), t(2;3)(q35;q21), t(2;3)(q33;q21), t(3;4)(p13;p16), t(3;6)(p13;q25), t(3;6)(q12;q15), t(3;6)(p23/24;q23/24), t(3;8)(p14;q24), t(3;9)(q21;p13), t(3;11)(q29;p15.3), t(3;12)(q13,q24), t(3;12)(p14;q13), t(3;14)(q21;q32), t(3;17)(p25;p13.3), t(3;21)(p12;q11.2). Gain of chromosome 7 (c-Met gene amplification) is associated with PRCC. Gain of chromosome 8 is linked to c-MYC gene.
(3) Kidney cancer is also a feature of the following syndromes:
The main inherited disorder predisposing to development CCRCC is von Hippel-Lindau (VHL) diease, which involves a germline mutation of the VHL gene at chromosome 3p25. Affected individuals are susceptible to tumors of multiple organ systems, including cysts and tumors of the kidney, which occur in 25-45% of cases with a mean age at onset of 40 years. The renal tumors are frequently multifocal and/or bilateral and are always of the CCRCC histological type.
Birt-Hogg-Dube syndrome (BHD) is another inherited disorder which predispose to multiple or bilateral RCC, particularly chromophobe and oncocytoma RCC, followed by papillary and clear cell RCC.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) or fumarate hydratase deficiency syndrome (FH) is mainly associated with papillary RCC.
Other familial symdromes that carry an increased risk for development of RCC include tuberous sclerosis complex (TSC) and hyperparathyroidism-jaw tumor syndrome (HRPT2). In addition, familial nonsyndromic RCC involves families in which multiple relatives develop RCCs taht often have the hallmarks of hereditary tumors (multifocality, bilaterality, and early age of onset) but for which no genetic cause has yet been discovered.
Diagnosis: In addition to a routine medical exam with a detailed medical history, MRIs, CT scans, ultrasounds, intravenous pyelograms, blood tests, urinalyses, and ateriography are used to diagnose kidney cancer.
Treatment: Treatment for kidney cancer depends on the type and stage of the disease. In wilms tumor, chemotherapy, radiotherapy and nephrectomy are the accepted treatments. In renal cell carcinoma, treatment depends on the stage of the cancer, but typically does not involve chemotherapy and radiotherapy, as renal cell carcinoma is resistant to these therapies. Treatment for localized renal cell carcinoma consists of nephrectomy alone, with no adjuvant (post surgical) therapy. In metastatic renal cell carcinoma,treatment consists of targeted therapies such as torisel, nexavar and sutent, the use of immunotherapy including interferon and interleukin-2, and in some cases, nephrectomy. Targeted cancer therapies such as sunitinib, temsirolimus, bevacizumab, interferon-alpha, and possibly sorafenib have improved the outlook for RCC (progression-free survival), although they have not yet demonstrated improved survival. Below is the kidney drugs approved by FDA.
Drugs Approved for Kidney (Renal Cell) Cancer.
For details about prevention and treatment of kidney cancer, click here.
Animal models:
(1) Nature models:
the Eker rat, a rodent model of hereditary cancer in which a single gene mutation predisposes them to bilateral multicentric renal cell carcinoma (Everitt et al. (1992). It was caused by a germline mutation in the tuberous sclerosis-2 (TSC2) gene (Kobayashi et al. 1995). The disorder bore similarities to von Hippel-Lindau disease. Rats that are heterozygous for the so-called Eker mutation develop spontaneous RCCs between 4 and 12 months of age. When homozygous, the mutation is lethal prenatally at 9 to 10 days of gestation. At the histologic level, renal carcinomas in the Eker rat develop through multiple stages from early preneoplastic lesions (e.g., atypical tubules) to adenomas in virtually all heterozygotes by the age of 1 year. Except for the occurrence of renal tumors, the phenotype of tuberous sclerosis in human differs from that of the Eker rat.
(2) Hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis (RCND) is a naturally occurring canine kidney cancer syndrome that was originally described in German shepherd dogs. Lingaas et al. (2003) narrowed the RCND interval to a small region on canine chromosome 5 that overlapped the human BHD gene. The authors described a his255-to-arg mutation in exon 7 of the canine Bhd gene that segregated with the disease phenotype.
(3) Nihon rat: Okimoto et al. (2004) reported a model of renal carcinoma found in a colony of Sprague-Dawley rats in Japan called 'Nihon 'rat. In heterozygotes, renal carcinomas developed from early preneoplastic lesions, seen as early as 3 weeks of age, into adenomas by 8 weeks of age with complete penetrance of this renal carcinoma gene by the age of 6 months. The renal carcinomas that developed in heterozygotes were predominantly of the clear cell type. Nihon rat bears a germline mutation in the BHD gene caused by insertion of a single nucleotide, resulting in a frameshift with a stop codon 26 amino acids downstream. They found that the homozygous mutant condition was lethal at an early stage of fetal life in the rat.
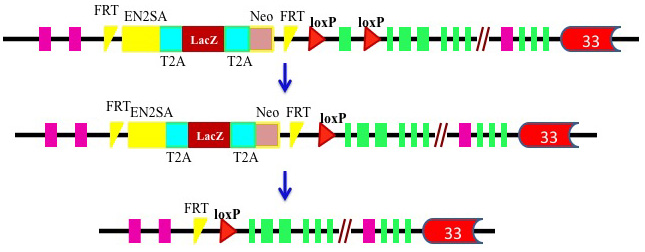
(4) Mouse models: Mice generated through genetic disruption of the kidney cancer-related genes Vhl, Met, Fh, Tsc1/Tsc2, Apc, or Pten, Bhd either do not develop RCC or present difficulties in studying renal tumorigenesis because of high neonatal mortality or long latency of tumor development and lack of tissue-specific targeting (Apc, Tsc-1, and Tsc-2). The inability of mouse models to recapitulate renal tumors has long puzzled kidney cancer researchers. Two groups have previously generated a kidney-specific knockout mouse model (Bhdflox/flox/Ksp-Cre) through disruption of the Bhd gene in the distal tubules, collecting ducts, and the descending limb of Henle’s loop (Babe et al, 2008, Chen et al, 2008). However, the affected mice rapidly developed polycystic kidney disease after birth and died in less than three weeks, probably due to disruption of Bhd in multiple types of kidney tubules. Recently, Chen et al. has developed a new kidney proximal tubule-specific Flcn knockout mouse model which develops a variety of kidney tumors, capitulating human BHD disease.
{kind=link}